PACSY (Protein structure And Chemical Shift NMR spectroscopY) is a relational database management system that integrates information from the Protein Database Bank (PDB), the Biological Magnetic Resonance Data Bank (BMRB), and the Structural Classification of Proteins (SCOP) database. To assist in structural investigations, PACSY provides three-dimensional coordinates and chemical shifts of atoms along with derived information such as torsion angles, solvent accessible surface areas, and hydrophobicities. PACSY consists of seven table types linked to one another for coherence by key identification numbers. Database queries are enabled by advanced search functions supported by an RDBMS server such as MySQL. The software packages PACSY Maker for database creation and PACSY Analyzer for database analysis are available from Programs tab. Both PACSY Maker and PACSY Analyzer are given in an executable form. PACSY Maker requires QT libraries to be executed. We provide only Linux version of PACSY Maker, however, generated SQL dump files can be moved to the other operating system to be installed. We provide Linux, Windows, and Mac versions of PACSY Analyzer.
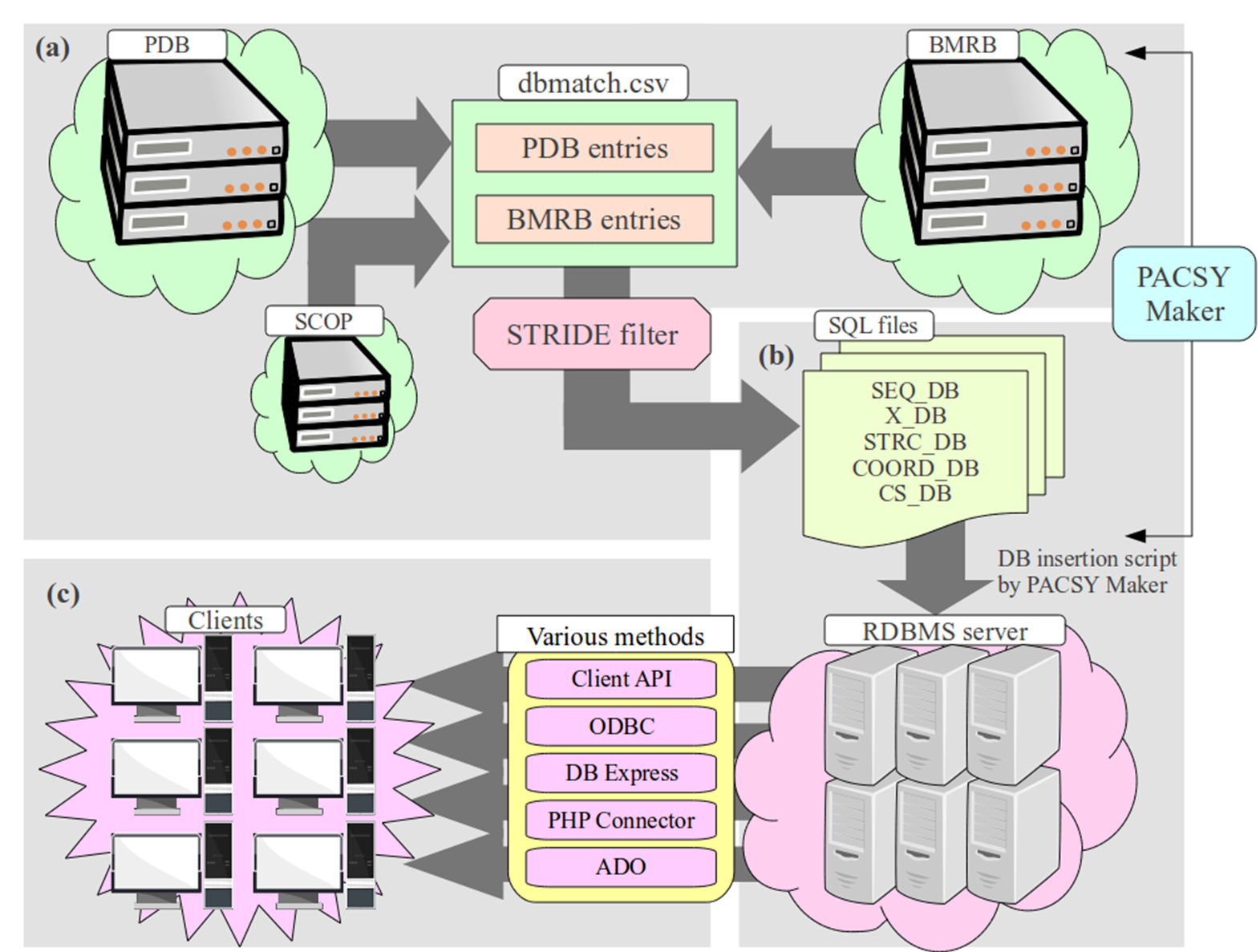
Table description
PACSY contains seven types of tables. Three types- SEQ_DB , SCOP_DB and MOLPROB_DB- consist only of one table each, while the others consist 20 tables each representing one of the 20 standard amino acid types. PACSY Maker extracts and processes necessary information to fill these tables from PDB, BMRB and SCOP. STRIDE is used to calculate secondary structure, solvent accessible surface areas (SASs) , and the hydrophobicities of the SASs. The separation of table types avoids storage of repetitive information (known as data anomalies). The “X” in front of a table type, stands for one of the 20 standard amino acids. Thus tables, X_DB, X_STRC_DB, X_CS_DB and X_COORD_DB are each actually 20 tables. Each type of table has a KEY_ID field. Thus, if chemical shift information about a certain residue is requested, it can be performed by querying both the X_CS_DB and X_DB with same KEY_ID. Whereas other table types each consist of 20 amino-acid-specific tables, SEQ_DB and SCOP_DB are single tables. They also have a KEY_ID field, whose value matches that of the X_CS_DB for the first residue of protein sequence.
| Table types | # of tables | # of fields | # of records | Description |
| PDBSEQ_DB | 1 | 6 | 291,344 | contains PDB sequence information regardless of experiment method. |
| SEQ_DB | 1 | 17 | 6,321 | contains basic information for entries. e.g. sequence, pH, temp, etc. |
| SCOP_DB | 1 | 10 | 236,955 | contains information of structural classification of proteins (SCOP). |
| MOLPROB_DB | 1 | 12 | 93,438 | contains information of structural quality of proteins assessed by MolProbity. |
| X_DB | 20 | 7 | 472,304 | contains residue related information. e.g. chain ID, sequence ID, amino acid type. |
| X_STRC_DB | 20 | 9 | 8,535,580 | contains structural information for a residue. e.g. secondary structure, dihedral angles, hydrophobicity, SAS, # of model. |
| X_COORD_DB | 20 | 7 | 122,827,204 | contains coordinate information for an atom. |
| X_CS_DB | 20 | 5 | 4,185,850 | contains chemical shift information for an atom including ambiguities. |